当前,全球有数以亿计人正遭受着肿瘤、糖尿病、青光眼等重大疾病的困扰。然而,这些疾病的治疗面临一个严峻的瓶颈:我们严重缺乏源于机理创新的药物靶点和原创性的治疗方法。传统的生命科学研究依赖周期漫长、成本高昂的实验试错,且难以系统性地揭示复杂疾病的动态全貌,导致新的生物发现和药物研发的效率低下。为了突破这一困境,我们必须引入一场研究范式变革。
由DNA控制的生命的本质是数字,人工智能(AI)可以洞见高维数据的内在特征,揭示复杂生物系统。我们的愿景是:构建"人工智能虚拟细胞"(AI Virtual Cell, AIVC)。这个愿景的核心:是利用前沿的AI技术,将海量的生命科学数据转化为一个可计算、可预测的in silico(在计算机中)生命模型。它将使我们从"解释已知"的研究模式,迈向至"预测未知"的新高度,从而将传统数年的研发周期缩短至数月,为攻克重大疾病提供原创发现与技术支撑。
我们的方法
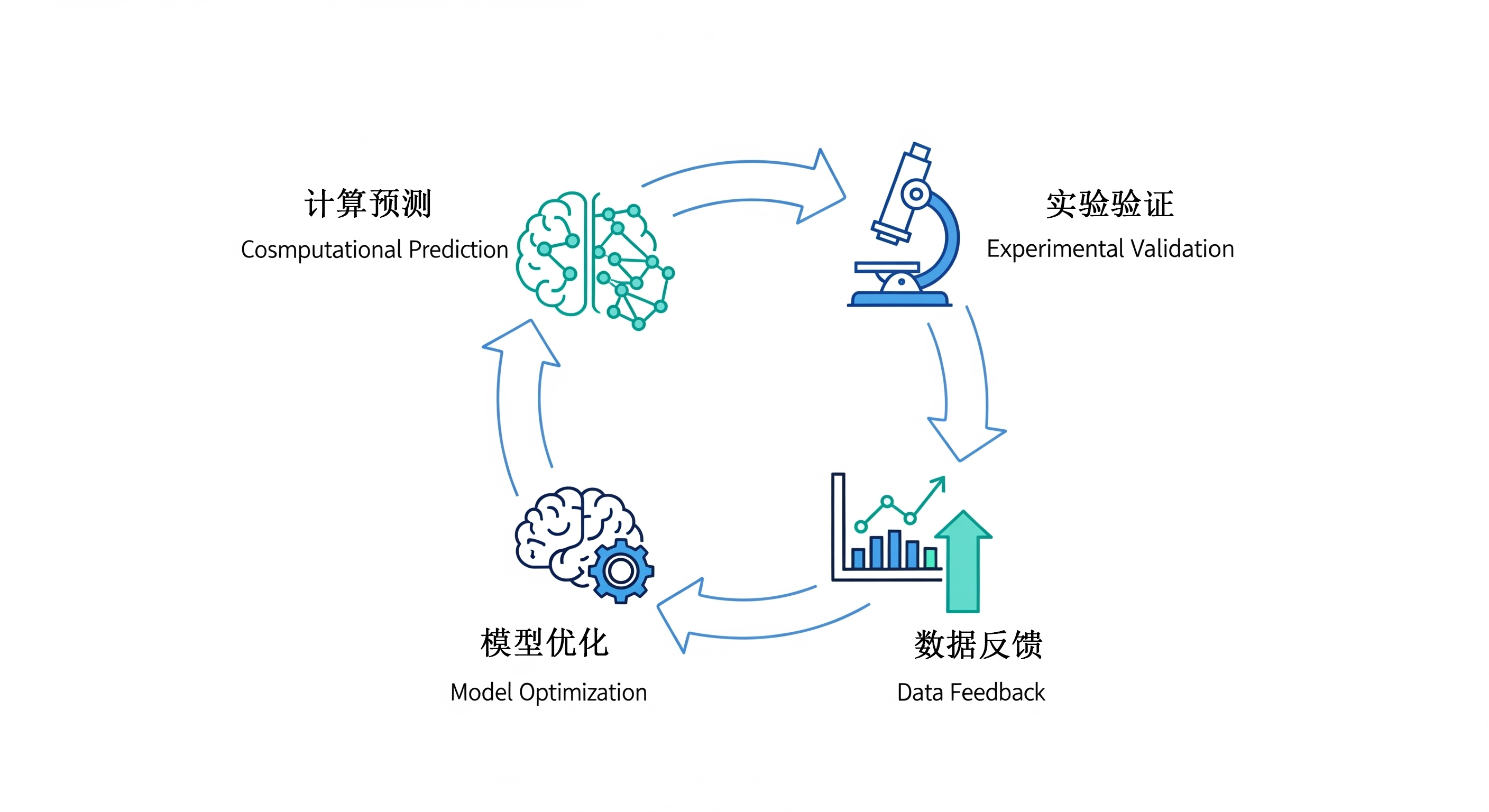
我们开展以计算生物学(干实验)和实验生物学(湿实验)紧密结合、双轮驱动的研究模式。我们相信,只有将前沿的计算分析与严谨的实验验证相结合,才能真正实现从海量数据到生物学机理、再到新型疗法的跨越。
我们是谁
我们的实验室汇集了来自不同领域的人才,创造了独特的跨学科环境。这种跨学科方法使我们能够从多个角度应对复杂挑战,推动创新和突破性发现。
- 机器学习专家:开发和应用先进的AI算法
- 生物信息学家:分析复杂的生物数据并整合多组学信息
- 分子生物学家:提供对细胞和分子机制的深入洞察
- 医学研究人员:确保我们的工作转化为具有临床相关性的应用
我们过去的研究
我们致力于构建从"AI算法与平台创新 → 功能机制解析 → 新型疗法开发"的完整研究链条。
发展了系列原创AI算法与计算平台,为解析复杂生命数据提供了新方法
现代生命科学带来了海量的多模态生物组学数据,我们围绕分子生物学中心法则里"基因、转录、翻译"三个核心基因调控层面,开发了一系列前沿算法和平台来攻克多个从数据处理到生物学洞察的关键技术瓶颈。
- 基因组学: 我们开发了高效的三代测序数据比对、纠错及拼装工具MECAT (Nature Methods, 2017)。在此基础上,我们系统性地评估了三代测序的变异检测工具 (Briefings in Bioinformatics, 2020),开发了新的基因组结构变异识别工具 (Genome Biology, 2021),并建立了领域首个基于三代测序数据的DNA甲基化数据库 (Nucleic Acids Research, 2017)。我们还构建了高质量的中国人群图形泛基因组 (Nature Communications, 2021),揭示了结构变异对人类表型和疾病的广泛影响 (Nucleic Acids Research, 2024)。
- 转录组学: 我们基于深度学习算法,开发了单细胞转录组数据填充算法DISC (Genome Biology, 2020),识别空间功能区域的新算法SECE (Biomolecules, 2024),以及能够整合空间转录组数据进行三维重构的STAIR算法 (Genome Biology, In review),为精准描绘细胞图谱奠定了方法学基础。
- 翻译组学: 我们创建和并持续更新领域广泛使用的 mRNA翻译组数据库RPFdb (Nucleic Acids Research, 2025; Nucleic Acids Research, 2019; Nucleic Acids Research, 2015),创建了首个可翻译环状RNA综合数据库riboCIRC (Genome Biology, 2021),并开发了基于自然语言交互的翻译组分析工具RiboChat (Briefings in Bioinformatics, 2022)。此外,我们系统总结了翻译组领域的计算工具和数据库资源 (Briefings in Bioinformatics, 2017),为领域内mRNA研究提供了核心数据资源和便捷的分析平台。
应用多组学技术系统揭示疾病和重要生物学机制
我们利用多种组学技术和生物信息学算法,对多种人类疾病,特别是眼科疾病,以及关键生物学过程进行了深度解析。
- 眼科及疾病机制: 针对视网膜母细胞瘤(RB),我们首次应用三代测序结合二代技术,全面鉴定了其致病性遗传变异 (Cancer Letters, 2024),并通过单细胞测序系统刻画了其恶性表型和肿瘤微环境的动态改变 (Cell Death and Disease, 2022)。我们发现了隐眼的致病基因并解析其生物学机制 (Human Molecular Genetics, 2018)。我们的研究体系同样展现了强大的跨领域应用潜力,在与顶尖肿瘤免疫专家的合作中,我们深入解析了错配修复缺陷型结直肠癌的免疫微环境,揭示了PD-1阻断疗法的重塑机制 (Cancer Cell, 2023),并发现了能够精准预测治疗反应的关键生物标志物 (Clinical Cancer Research, 2024),展现了我们研究体系在重大疾病临床转化中的价值。
- 视觉系统发育和衰老: 我们系统描绘了小鼠视网膜发育过程中的RNA编辑 (BMC Biology, 2024) 和翻译调控的时空全景图 (Nucleic Acids Research, 2021),并利用空间转录组技术解析了衰老对大脑功能区域,特别是视觉皮层的影响 (Aging Cell, 2024)。
- mRNA翻译生物调控: 我们开创性地揭示了核糖体蛋白缺失如何系统性地重塑细胞转录和翻译景观,并重点解析了核糖体蛋白在新生血管生成以及视网膜细胞命运决定的功能 (Nucleic Acids Research, 2022、封面论文、重大突破论文)。此外,我们首次系统绘制和比较了小鼠多组织、多发育阶段的翻译组图谱 (Nucleic Acids Research, 2021)。
开创mRNA等新型疗法在神经保护的应用,并验证了其巨大潜力
我们将基础研究的见解转化为新型治疗策略的开发,特别是在mRNA药物领域取得了关键突破。
- 神经损伤保护: 我们开发了高效的无疤痕环状RNA(circRNA)系统,作为一种提升蛋白表达效率和稳定性的新型RNA疗法平台 (Molecular Therapy Nucleic Acids, 2025)。针对视网膜神经节细胞(RGC)损伤后难以再生这一重大挑战,我们证实了mRNA疗法能够为RGC提供持续且强大的神经保护作用,这也是利用可翻译RNA治疗眼科及神经退行性疾病的开创性工作之一 (Molecular Therapy-Nucleic Acids, 2024、封面论文)。此外,我们参与了冬眠动物的诱导多能干细胞研究项目,为研究视网膜等组织如何适应极端低温环境及其在神经保护等领域的医学应用提供了全新的视角 (Cell, 2018、封面论文)。
- AI赋能mRNA药物设计: 针对mRNA药物翻译效率低下的核心难题,我们开创性地开发了基于深度生成模型的mRNA密码子优化平台RiboDecode。该平台能够智能生成兼具高蛋白表达效率和功能性的mRNA序列,动物实验证明其有效性:在RGC损伤模型中,经我们平台优化的mRNA药物仅用1/5的剂量,即达到与未优化药物同等的神经保护效果 (Nature Communications, In revision)。这也是首次在动物实验里证实,AI生成的mRNA序列可以极大提升其蛋白替代疗法的治疗潜力。
与我们合作
我们一直在寻找对AI、生物信息学、分子与细胞生物学和再生医学充满热情的人才。如果您有兴趣加入我们的多学科团队或与我们合作,请与我们联系(xiezhi@gmail.com)。